Simulation Results Display
Molecular Structure and Adsorption Analysis
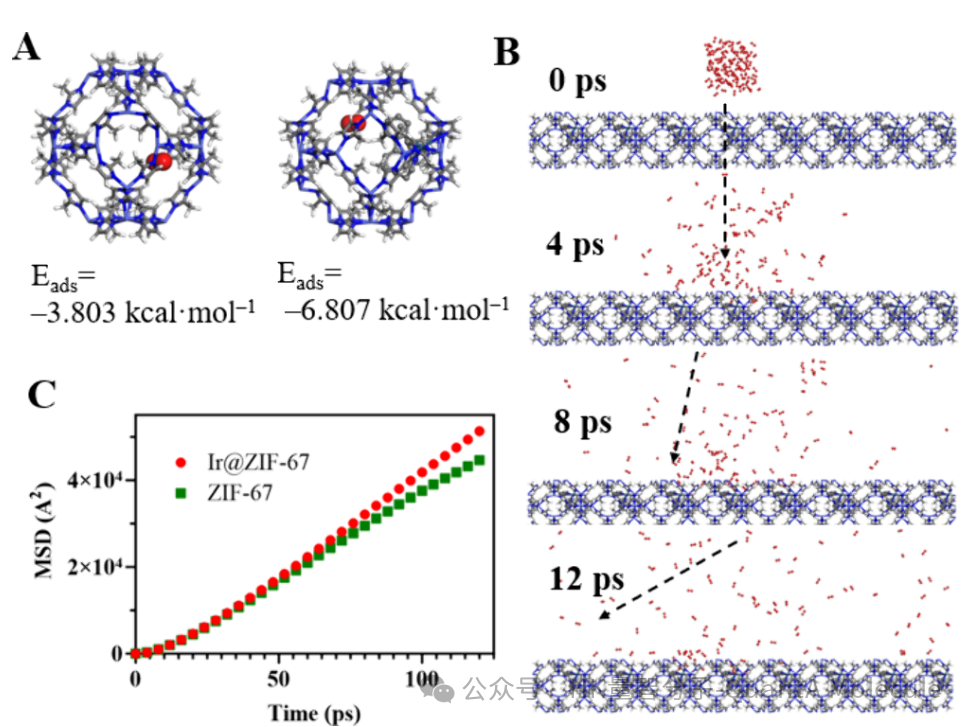
The figure above shows the adsorption behavior of molecules in Metal-Organic Framework (MOF) materials. By calculating the binding energy (Eads) at different adsorption sites, we can predict the main adsorption positions and strengths of molecules in the material. The binding energies of two different adsorption sites in the figure are -3.803 kcal·mol-1 and -6.807 kcal·mol-1, indicating that the red molecule has stronger binding ability at the right site.
These adsorption data are crucial for understanding the initial state and diffusion starting point of molecules in materials, directly affecting the calculation results of subsequent diffusion paths and energy barriers.
Diffusion Trajectory and MSD Analysis
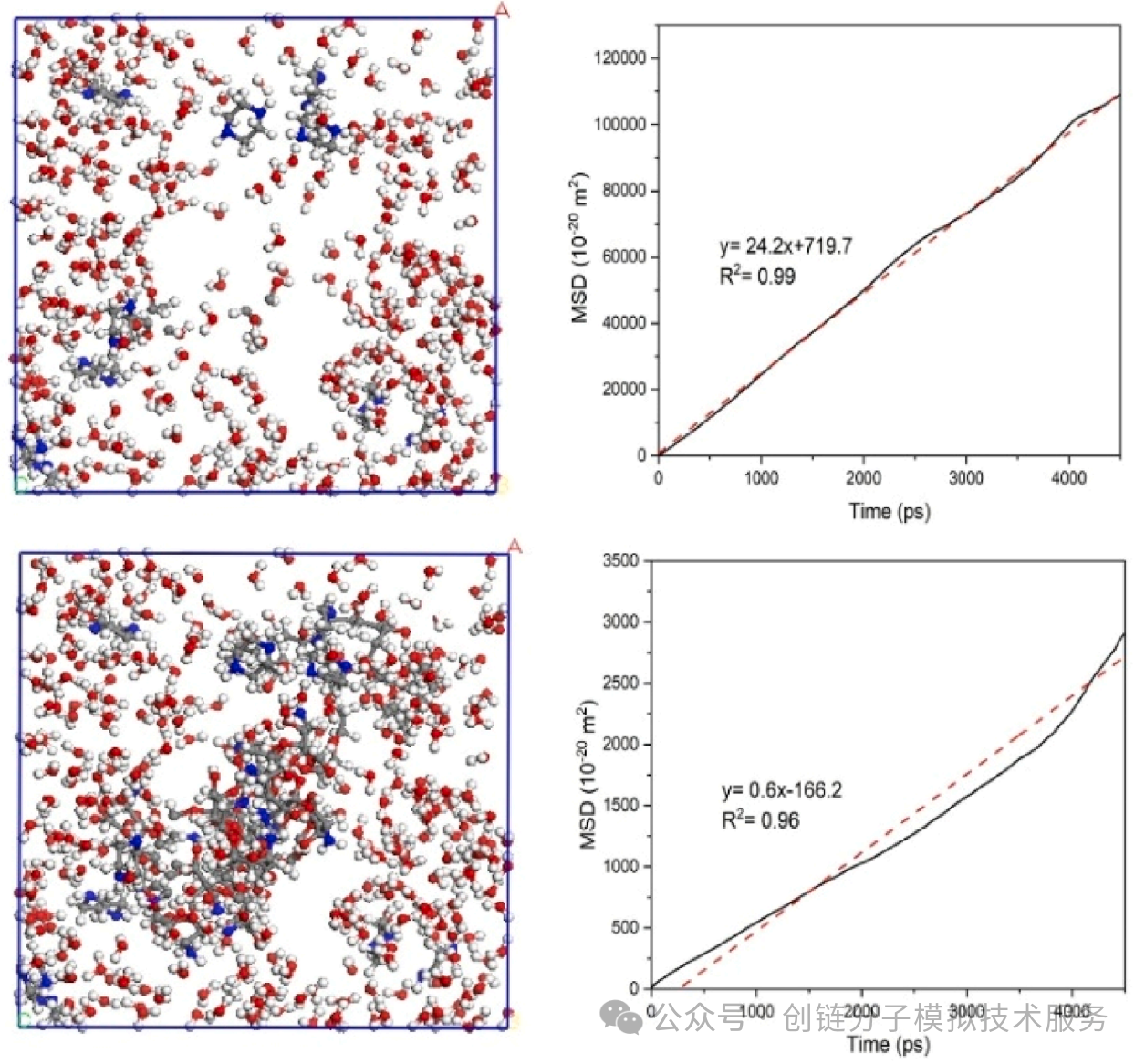
The right part of the figure shows the diffusion trajectory of molecules at different time points (0 ps, 4 ps, 8 ps, 12 ps), clearly presenting how molecules migrate through material channels. The left MSD (Mean Square Displacement) curve provides quantitative analysis, with the red curve representing ZIF-67 material loaded with Ir metal and the green curve representing pure ZIF-67 material.
It can be seen from the MSD curve that the molecular diffusion rate in Ir-loaded materials is significantly higher than that in pure materials, indicating that metal modification can effectively improve the transport properties of materials. The diffusion coefficient can be calculated through the slope of the MSD curve, providing important basis for material performance optimization.
