模拟结果展示
分子结构与吸附分析
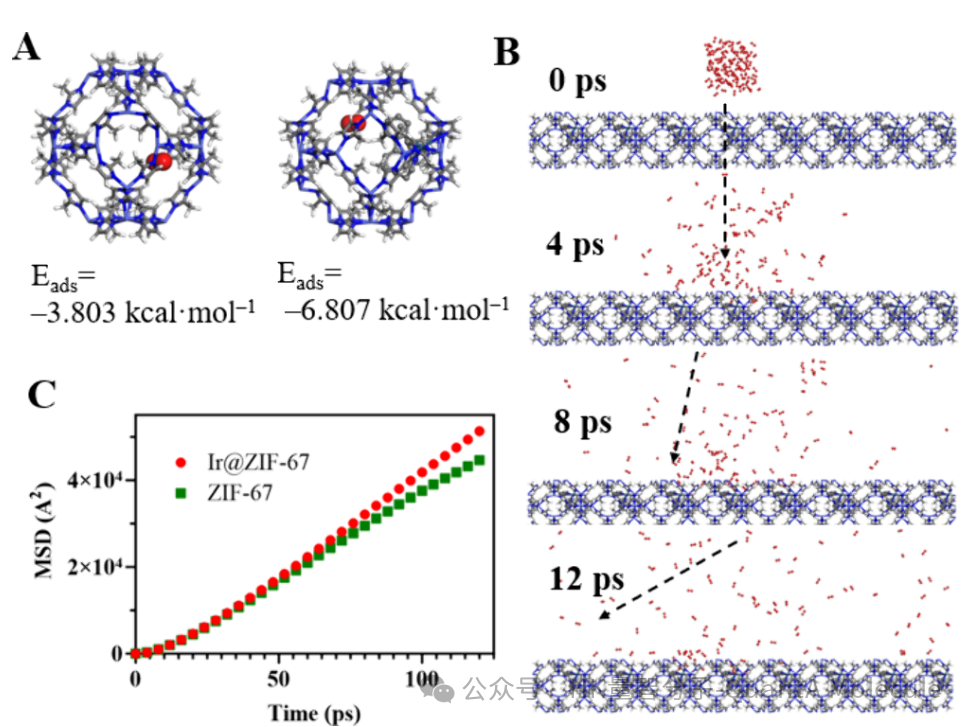
上图展示了金属有机框架(MOF)材料中分子的吸附行为,通过计算不同吸附位点的结合能(Eads),可以预测分子在材料中的主要吸附位置和吸附强度。图中两个不同吸附位点的结合能分别为-3.803 kcal·mol-1和-6.807 kcal·mol-1,表明红色分子在右侧位点具有更强的结合能力。
这些吸附数据对于理解分子在材料中的初始状态和扩散起点至关重要,直接影响后续扩散路径和能垒的计算结果。
扩散轨迹与MSD分析
上图右侧部分展示了分子在不同时间点(0 ps、4 ps、8 ps、12 ps)的扩散轨迹,清晰地呈现了分子如何通过材料孔道进行迁移。左侧MSD(均方根位移)曲线则提供了定量分析,其中红色曲线代表负载金属Ir的ZIF-67材料,绿色曲线代表纯ZIF-67材料。
从MSD曲线可以看出,负载Ir的材料中分子扩散速率明显高于纯材料,这表明金属修饰可以有效提升材料的传输性能。通过MSD曲线斜率可以计算扩散系数,为材料性能优化提供重要依据。