吸附相关计算
精确计算不同条件下的吸附量和吸附位点,为材料设计和性能优化提供关键数据支持
技术概述
吸附相关计算是一种重要的分子模拟技术,用于研究材料表面与分子之间的相互作用。通过精确计算不同条件下的吸附量和吸附位点,可以深入理解吸附机制,为新型吸附材料的设计和性能优化提供重要理论依据。
该技术广泛应用于催化剂设计、气体分离、水处理、储能材料等领域的研发,可以有效预测材料的吸附性能,加速研发过程并降低实验成本。
竞争吸附分析
精确计算混合组分在不同比例下的竞争吸附行为,评估材料的选择性和分离性能
吸附位点识别
识别和分析材料表面的吸附位点,计算不同位点的吸附能和结合强度
吸附量计算
吸附量是评估吸附材料性能的关键指标,包括混合组分不同比例竞争吸附、混合组分饱和吸附、单组分饱和吸附等多种情况。通过分子模拟,我们可以精确计算不同条件下的吸附量数据。
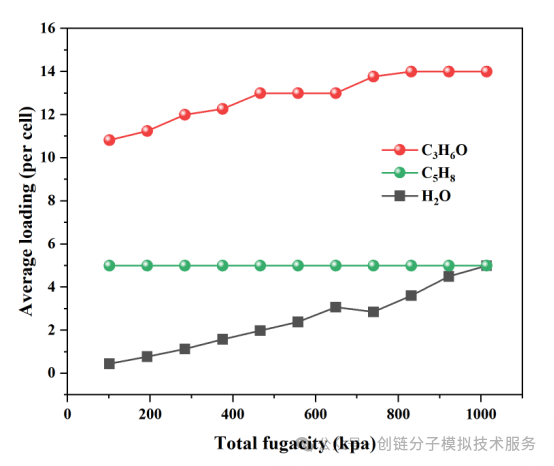
吸附性能数据
吸附性能数据表提供了不同材料在标准条件下的详细吸附参数,包括吸附容量、选择性系数、吸附热等关键指标,为材料筛选和性能评估提供重要参考。
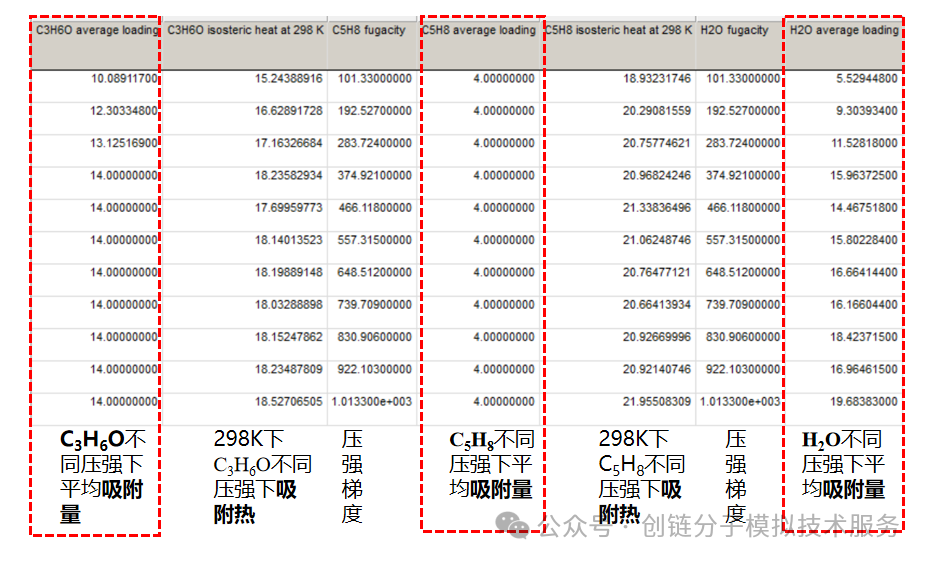
吸附位点分析
吸附位点分析是理解吸附机制的重要手段。通过分子模拟,我们可以直观观察分子在材料表面的吸附位置和结合方式,计算不同位点的吸附能,为材料设计提供分子层面的指导。
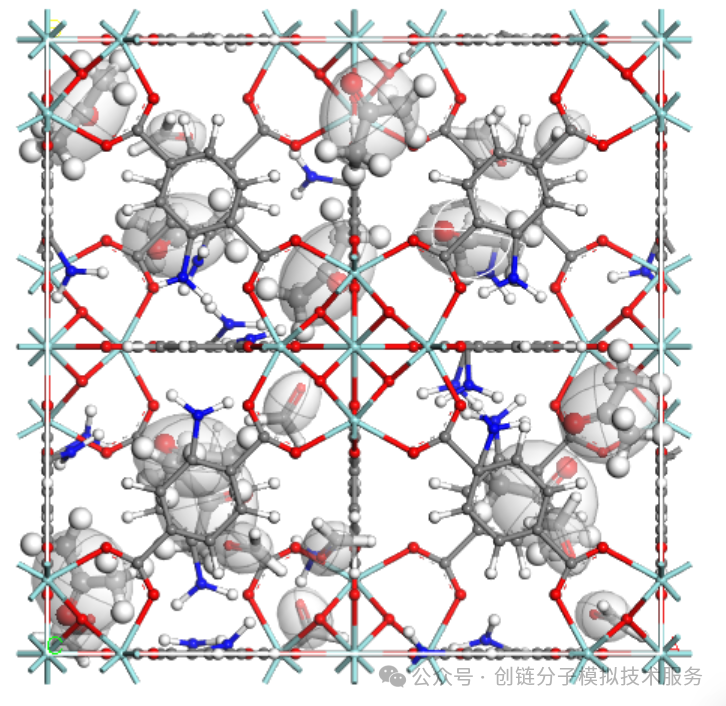
分析方法
通过密度泛函理论(DFT)计算和分子动力学模拟,我们可以系统分析吸附位点的以下特征:
- 吸附位点的几何结构和电子环境
- 不同吸附位点的能量分布和稳定性
- 分子在不同位点的吸附构型和结合方式
典型应用案例
CO2捕获材料设计
模拟不同金属氧化物和碳材料的CO2吸附性能,优化材料组成和制备条件,开发高效的CO2捕获材料。
气体分离膜优化
研究混合气体在膜材料中的竞争吸附行为,设计具有高选择性和高通量的气体分离膜。
催化材料活性位点识别
识别催化剂表面的活性吸附位点,分析反应物在活性位点的吸附状态,为提高催化效率提供理论指导。
技术参数
- 模拟温度范围 273-1000 K
- 压力范围 0.01-10 MPa
- 吸附组分数量 1-10种
- 吸附位点计算精度 meV级
- 吸附等温线类型 Langmuir, Freundlich, Toth等
相关技术
- 分子动力学模拟(压力场)
- 分子动力学模拟(电场)
- 自由扩散模拟
- 分子结构性能计算