Material Structure Property Calculation
High-precision quantum chemistry calculations, in-depth analysis of molecular electronic structure, reaction activity and thermodynamic properties, providing theoretical guidance for material design and drug development.
Key Analysis Items for Material Structure Property Calculation
1. Molecular Structure Optimization and Geometric Parameters
Molecular structure optimization is the fundamental step in quantum chemistry calculations, determining the stable geometric configuration of molecules through energy minimization. The optimized structure includes key geometric parameters such as bond lengths, bond angles, and dihedral angles, which are important foundations for understanding molecular properties.
We use high-precision DFT methods (such as B3LYP/6-31G(d,p)) for geometric optimization to ensure the accuracy and reliability of calculation results. The optimized structures can be used for subsequent electronic structure analysis, spectroscopy calculations, and other advanced studies.
2. HOMO-LUMO Orbital Analysis
Frontier orbital (HOMO-LUMO) theory is an important tool for understanding molecular reactivity. The energy difference between HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) determines a molecule's electron transition capability and chemical reactivity.
By analyzing the energy and shape of frontier orbitals, we can predict a molecule's nucleophilic/electrophilic reaction sites, light absorption properties, and electron transport characteristics. The HOMO-LUMO energy gap (such as 3.07 eV) is a key parameter for evaluating molecular conductivity and optical properties.
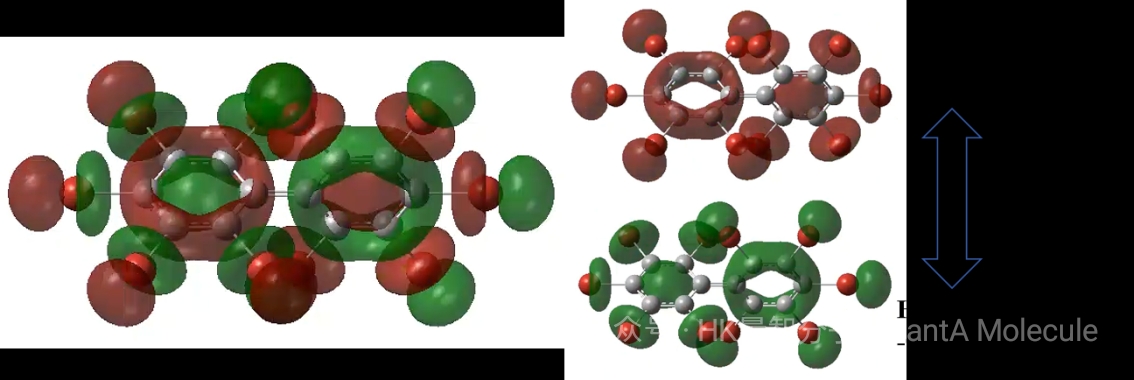
3. Differential Charge Density Calculation
Differential charge density maps visually show the redistribution of electron density in molecules, with red regions indicating increased electron density and blue regions indicating decreased electron density. This is crucial for understanding chemical bond polarity, electron transfer, and intramolecular interactions.
Through differential charge density analysis, we can quantitatively evaluate the amount of charge transfer (such as 0.32 e), identify polar covalent bond characteristics, and understand intramolecular conjugation effects. This information is of great guiding significance for designing molecular materials with specific electronic properties.
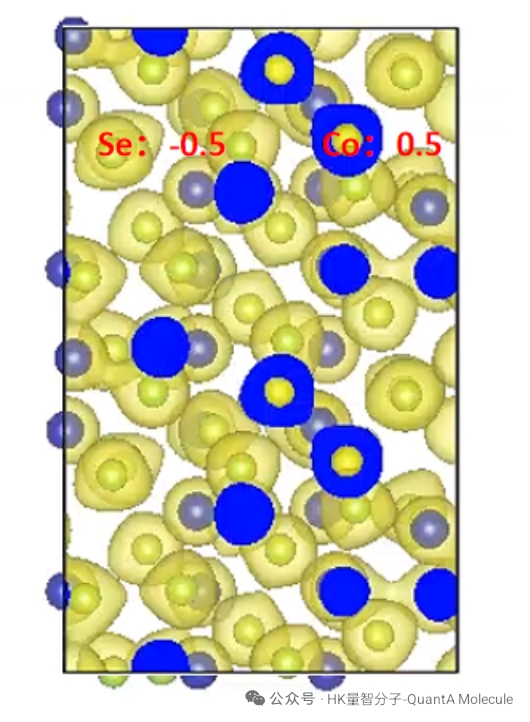
4. Bader Charge Calculation
Bader charge analysis is based on the topological properties of electron density and can accurately partition atomic charges in molecules. This method considers the actual distribution of electron density and is more accurate and reliable than traditional Mulliken charge analysis.
Bader charge results can reveal atomic oxidation states, electron affinity, and ionic characteristics of chemical bonds. This information is of great value for understanding catalytic reaction mechanisms, molecular recognition, and material electronic properties.
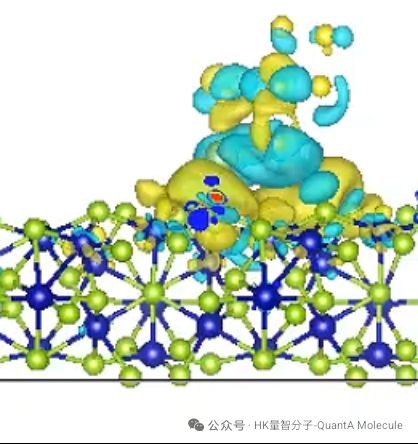
5. Electrostatic Potential Calculation
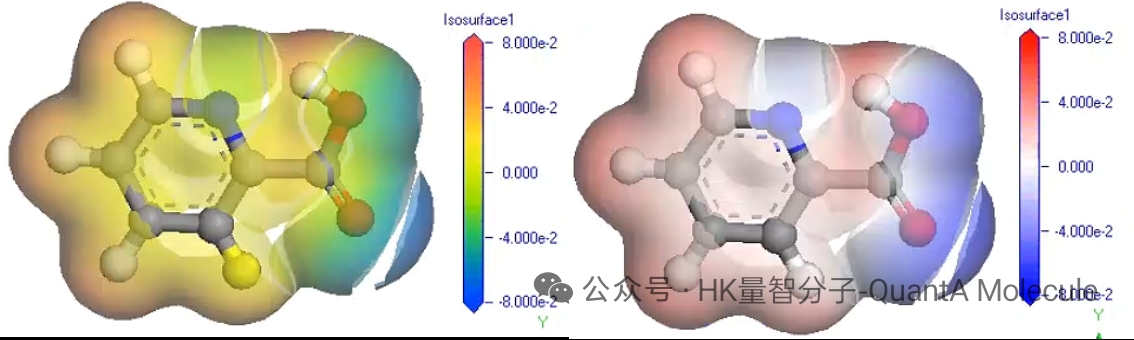
Molecular electrostatic potential maps describe the charge distribution on the molecular surface and can visually show the nucleophilic/electrophilic sites of molecules. Red regions indicate negative potential (electron-rich areas), and blue regions indicate positive potential (electron-deficient areas).
Electrostatic potential analysis is of great significance for predicting intermolecular interactions, solvation effects, and reaction sites. By studying the electrostatic potential distribution on the molecular surface, we can optimize molecular design to enhance specific interaction capabilities.
6. Density of States (DOS) and Projected Density of States (PDOS) Analysis
Density of states analysis provides information about the energy distribution of molecular electronic structure. Total density of states (DOS) shows the distribution of electronic states at different energies, while projected density of states (PDOS) further decomposes the contribution of different atomic orbitals to electronic states.
Through DOS and PDOS analysis, we can determine the bonding characteristics of molecules, hybrid orbital components, and the contributions of different atoms in specific energy regions. This is crucial for understanding molecular electronic structure and designing materials with specific electronic properties.
、分波态密度(PDOS).jpg)
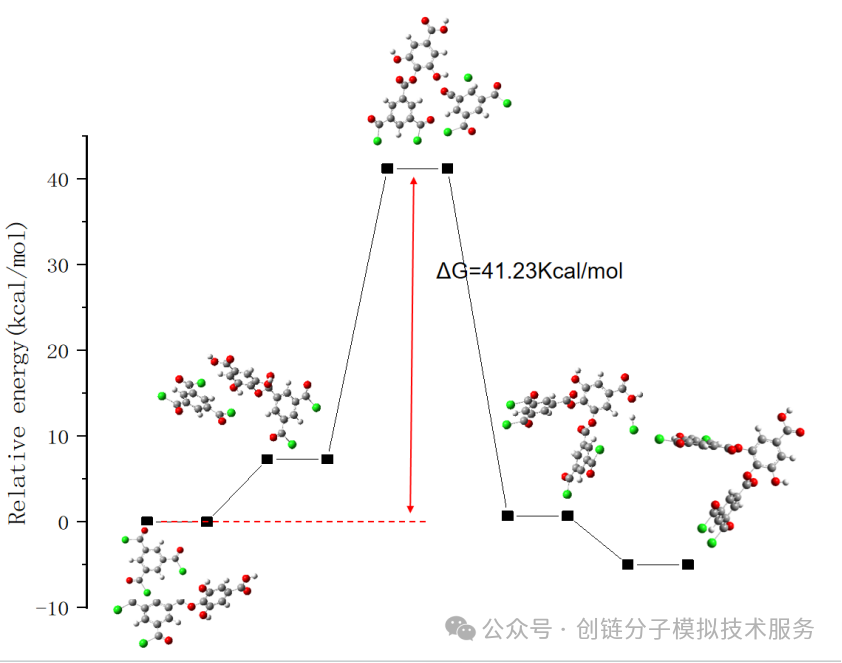
7. Thermodynamic Property Calculation (Gibbs Free Energy, Zero-Point Energy)
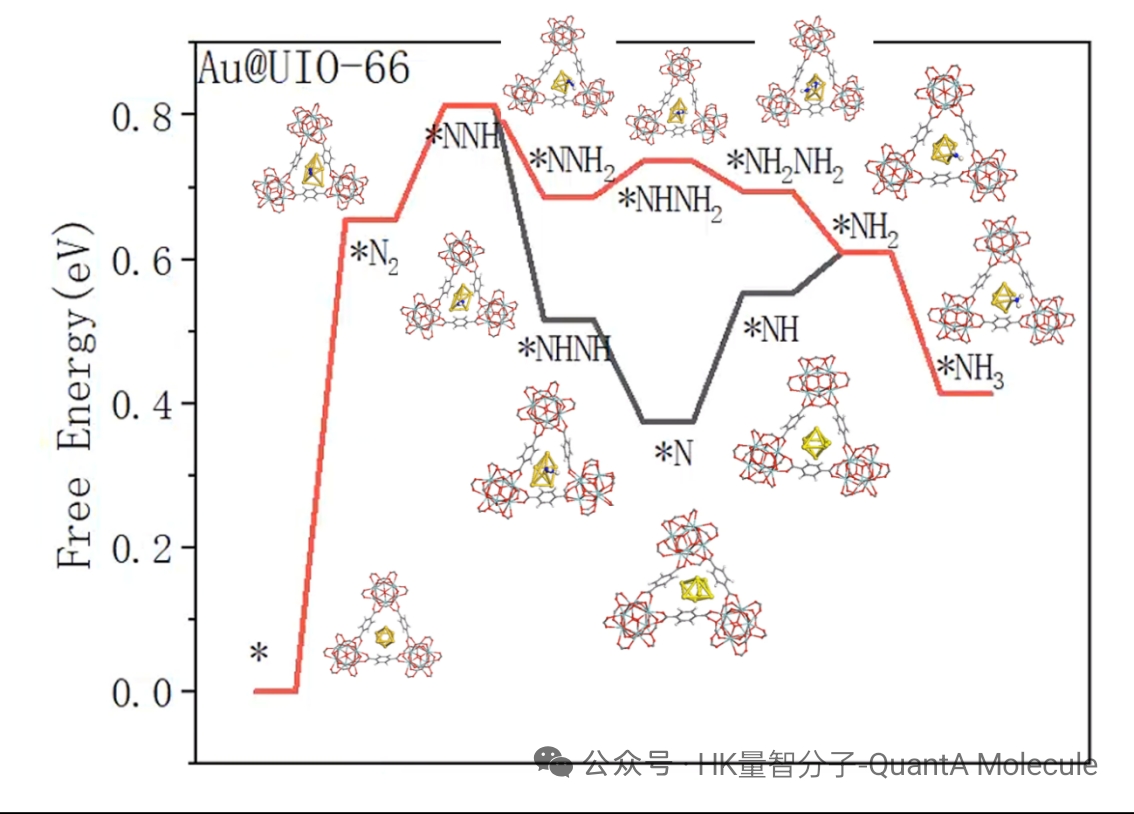
Thermodynamic property calculations include parameters such as total energy, enthalpy, entropy, Gibbs free energy, and zero-point energy (ZPE). These properties are crucial for evaluating molecular stability, reaction thermodynamics, and phase equilibrium behavior.
Through thermodynamic data obtained from frequency calculations, we can predict the spontaneity of reactions (judged by ΔG), calculate equilibrium constants and reaction rates. Zero-point energy correction is particularly important for accurately predicting reaction energy changes, especially in reactions involving hydrogen transfer.
Technical Advantages of Material Structure Calculations
High-Precision Calculation
Advanced quantum chemistry methods and basis sets ensure the accuracy and reliability of calculation results, providing reliable theoretical predictions for experiments.
Molecular-Level Insights
Provides molecular-level information difficult to directly observe through experiments, deeply understanding molecular electronic structure, reaction mechanisms, and property relationships to guide material design.
Efficient R&D
Quickly screen candidate molecules, predict their properties, significantly shorten R&D cycles, reduce experimental costs, and accelerate the development of new materials and drugs.
Main Application Fields
Drug Molecular Design
Predict interactions between drug molecules and targets, optimize molecular structures to improve activity and selectivity
Catalyst Design
Analyze catalytic active sites, predict reaction pathways and activity, design efficient catalysts
New Material Development
Predict electronic, optical and thermodynamic properties of new functional materials, guide material design
Green Chemistry
Design environmentally friendly chemical reaction pathways, reduce energy consumption and pollutant emissions
Need Professional Material Structure Calculation Services?
Our expert team can provide you with customized quantum chemistry calculation services to support your material research and drug innovation.