分子结构性能计算
高精度量子化学计算,深入分析分子电子结构、反应活性和热力学性质,为材料设计和药物研发提供理论指导。
分子结构性能计算关键分析项目
1. 分子结构优化与几何参数
分子结构优化是量子化学计算的基础步骤,通过能量最小化确定分子的稳定几何构型。优化后的结构包含键长、键角、二面角等关键几何参数,是理解分子性质的重要基础。
我们使用高精度DFT方法(如B3LYP/6-31G(d,p))进行几何优化,确保计算结果的准确性和可靠性。优化后的结构可用于后续的电子结构分析、光谱计算等高级研究。
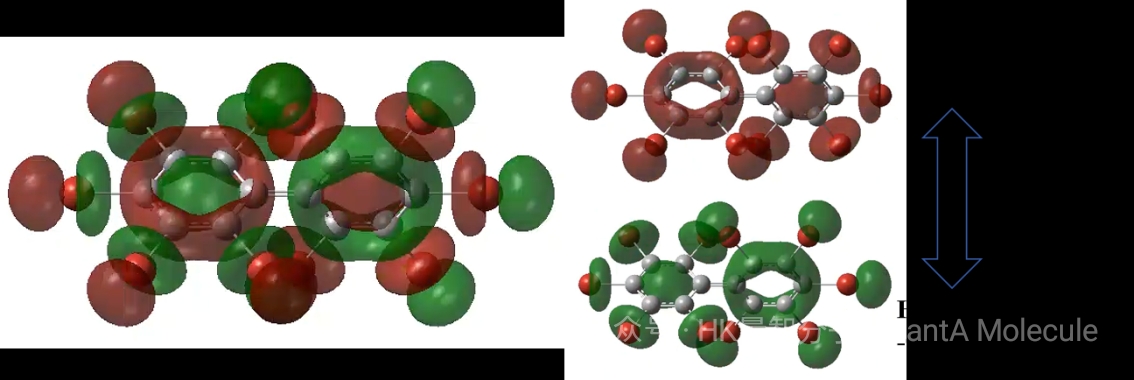
2. HOMO-LUMO轨道分析
前线轨道(HOMO-LUMO)理论是理解分子反应活性的重要工具。HOMO(最高占据分子轨道)和LUMO(最低未占据分子轨道)的能量差决定了分子的电子跃迁能力和化学反应活性。
通过分析前线轨道的能量和形状,可以预测分子的亲核/亲电反应位点、光吸收性质和电子传输特性。HOMO-LUMO能隙(如3.07 eV)是评估分子导电性和光学性能的关键参数。
3. 差分电荷密度计算
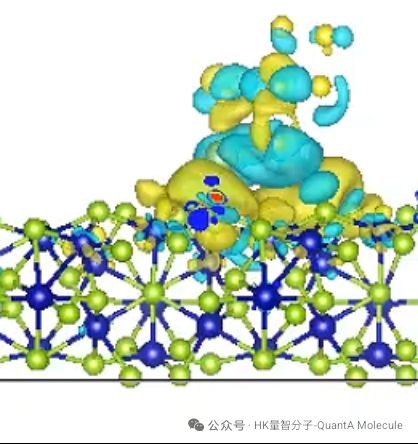
差分电荷密度图直观展示了分子中电子密度的重新分布,红色区域表示电子密度增加,蓝色区域表示电子密度减少。这对于理解化学键的极性、电子转移和分子内相互作用至关重要。
通过差分电荷密度分析,可以定量评估电荷转移量(如0.32 e),识别极性共价键特征,并理解分子内共轭效应。这些信息对于设计具有特定电子性质的分子材料具有重要指导意义。
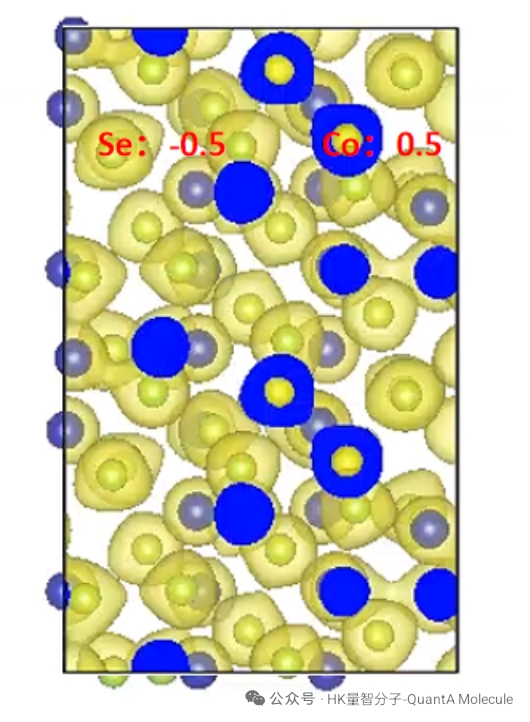
4. Bader电荷计算
Bader电荷分析基于电子密度的拓扑性质,能够准确划分分子中的原子电荷。该方法考虑了电子密度的实际分布,比传统的 Mulliken 电荷分析更为准确和可靠。
Bader电荷结果可以揭示原子的氧化态、电子亲和性和化学键的离子特性。这些信息对于理解催化反应机制、分子识别和材料电子性质具有重要价值。
5. 静电势计算
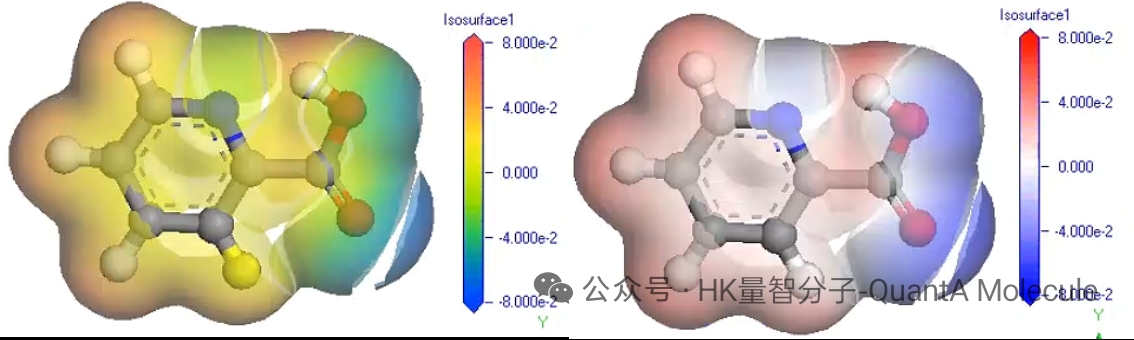
分子静电势图描述了分子表面的电荷分布情况,可以直观展示分子的亲核/亲电位点。红色区域表示负电势(电子富集区),蓝色区域表示正电势(电子亏损区)。
静电势分析对于预测分子间相互作用、溶剂化效应和反应位点具有重要意义。通过研究分子表面的静电势分布,可以优化分子设计以增强特定的相互作用能力。
6. 态密度(DOS)和分波态密度(PDOS)分析
态密度分析提供了分子电子结构的能量分布信息。总态密度(DOS)显示了电子态在不同能量处的分布,而分波态密度(PDOS)则进一步分解了不同原子轨道对电子态的贡献。
通过DOS和PDOS分析,可以确定分子的成键特性、杂化轨道成分以及不同原子在特定能量区域的贡献。这对于理解分子的电子结构和设计具有特定电子性质的材料至关重要。
、分波态密度(PDOS).jpg)
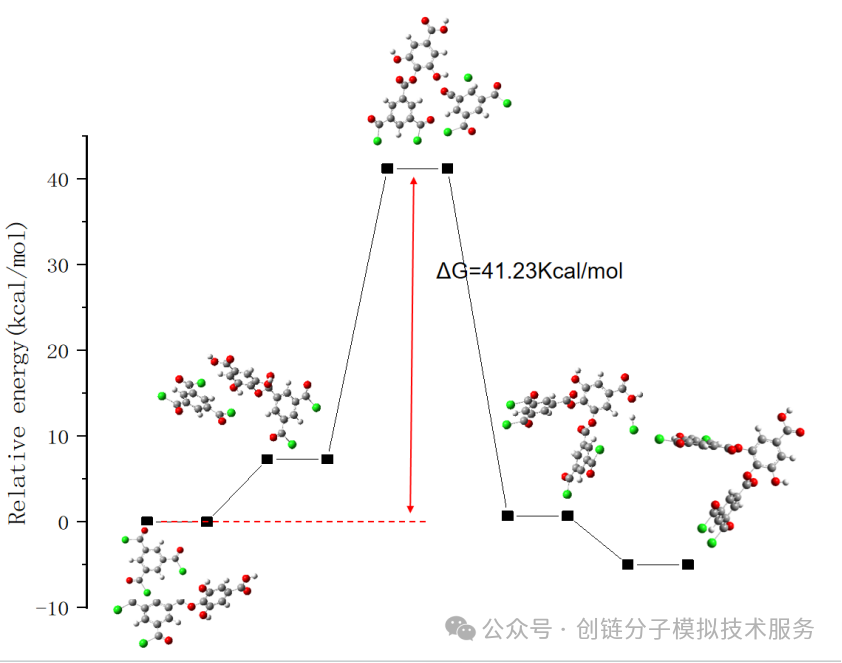
7. 热力学性质计算(Gibbs自由能、零点能)
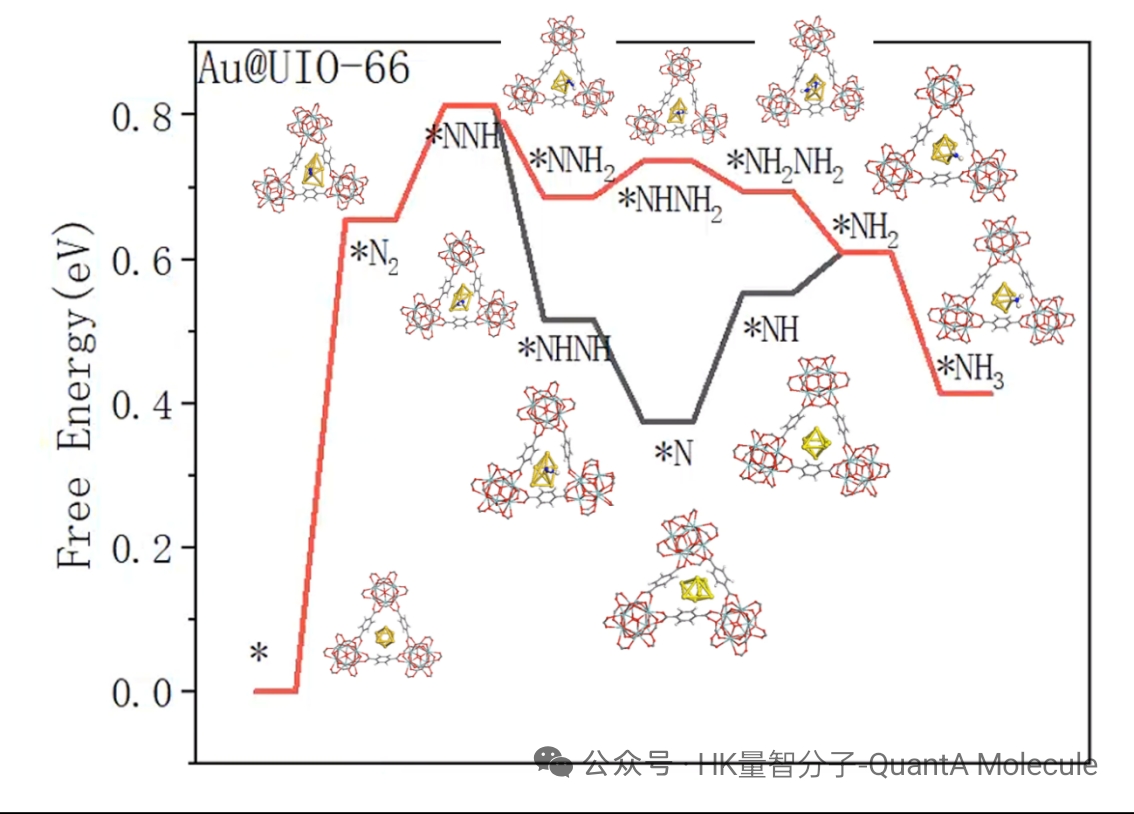
热力学性质计算包括总能量、焓、熵、Gibbs自由能和零点能(ZPE)等参数。这些性质对于评估分子的稳定性、反应热力学和相平衡行为至关重要。
通过频率计算获得的热力学数据,可以预测反应的自发性(通过ΔG判断)、计算平衡常数和反应速率。零点能矫正对于准确预测反应能量变化尤为重要,特别是在涉及氢转移的反应中。
分子结构计算的技术优势
高精度计算
采用先进的量子化学方法和基组,确保计算结果的准确性和可靠性,为实验提供可靠的理论预测。
分子级洞察
提供实验难以直接观测的分子级信息,深入理解分子的电子结构、反应机理和性能关系,指导材料设计。
高效研发
快速筛选候选分子,预测其性能,大幅缩短研发周期,降低实验成本,加速新材料和药物的开发进程。
主要应用领域
药物分子设计
预测药物分子与靶点的相互作用,优化分子结构以提高活性和选择性
催化剂设计
分析催化活性位点,预测反应路径和活性,设计高效催化剂
新材料开发
预测新型功能材料的电子、光学和热力学性能,指导材料设计
绿色化学
设计环境友好的化学反应路径,降低能耗和减少污染物排放